
Post Market Clinical Follow-up (PMCF) is a complex and often misunderstood element in the MedTech space. With Europe’s medical device regulation (EUMDR) kicking in, the manufacturers will need a more robust PMCF plan. We had the opportunity to attend a session on the topic: Updates and Strategies for Post Market Clinical Follow-up (PMCF), at the Virtual MedTech: EUMDR Exchange Conference, 2021. The session was conducted by Bassil Akra, CEO and Co-Owner, QUNIQUE.
This presentation focused on assessing what manufacturers need to do to perform compliant post market clinical follow-up (PMCF) activities and the best practices for completing PMCFs.
Assessing what manufacturers need to do to perform compliant PMCF activities
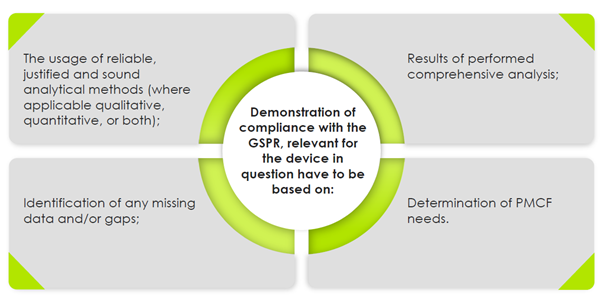
Manufacturers “of devices shall implement and keep up to date the post-market surveillance system” – Article 10[10] of the Medical Devices Regulation [MDR] and Article 10[9] of the In Vitro Diagnostic Regulation [IVDR]. “The manufacturer shall specify and justify the level of clinical evidence necessary to demonstrate compliance with the relevant general safety and performance requirements which shall be appropriate to the characteristics of the device and its intended purpose.” – Annex I, MDR. In case of no clinical data, the manufacturers should collect PMCF data, where the PMCF plan is also deemed necessary.
The clinical development plan (CDP) indicates a progression from exploratory investigations, such as first-in-man studies and feasibility and pilot studies, to confirmatory investigations, such as pivotal clinical investigations, and a PMCF (Annex XIV, MDR). The MDCG 2020-6 can be referred for the analysis of these clinical data.
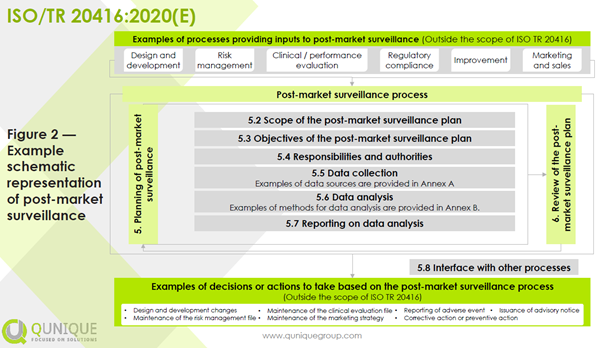
For any device, manufacturers shall plan, establish, document, implement, maintain, and update a post-market surveillance (PMS) system in a manner that is proportionate to the risk class and appropriate for the type of device throughout the lifetime of the device. Some regional regulatory requirements differ among the expected lifetime, entire lifetime, and lifetime of a device. The expected lifetime is to be defined during the design input phase by considering the current state of the art for a specific intended use and indication of a device. The entire lifetime is to be understood as the period from the first placing on the market after initial market approval till the last patient is treated with the device and followed-up for the predefined expected lifetime by the manufacturer. The lifetime of a device is to be understood as the validated expected lifetime according to real world evidence. A schematic representation of PMS is shown in the figure below:
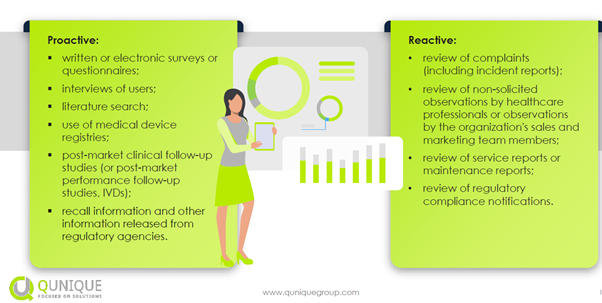
The proactive and reactive methods are shown in the figure below:
Best practices for completing PMCFs
PMCF is a continuous process to update the clinical evaluation and is part of the PMS plan. The PMCF plan includes the general methods and procedures of PMCF (i.e., gathering clinical experience, collecting feedback from users, screening of scientific literature, and of other sources of clinical data) along with the specific methods and procedures of PMCF to be applied (i.e., evaluation of suitable registries or PMCF studies). The MDCG 2020-7 is the guidance document for the PMCF plan template. The PMCF plan should link to the CER, connecting the methodologies selected and the methods employed.
The different procedures that are to be used as part of a PMCF include screening of scientific literature and other sources of clinical data, post-market studies, collecting data from registries, surveys of health care professionals, surveys of patients/users, and reviews of case reports that may reveal misuse or off-label use. The PMCF might not always be a study. For instance, reporting of a system misuse/off-label use cannot be found by a PMCF study, but it can rather be found via a survey or customer feedback. The rationale for the appropriateness of the chosen methods/procedures should also be discussed.
The specific timeline for the PMCF activities should be provided; a detailed and adequately justified time schedule for PMCF activities, such as the analysis of PMCF data and reporting, should be described. A summary table of the different PMCF activities foreseen by the manufacturer is provided below:

The applicable common specification(s), harmonized standard(s), or applicable guidance document(s) should be referenced, and the estimated date of the PMCF report should be mentioned in the PMCF plan. In addition, the relevant parts of the technical documentation should also be clearly referenced. The PMCF plan should align with the RMR and the clinical evaluation.
Conclusion
An ideal PMCF plan, with reference to MDCG 2020-7, presents the general and specific methods and procedures of the PMCF activities. The PMCF report should reflect as an output of such a PMCF plan and align with the instructions for use, clinical evaluation, summary of safety and clinical performance, and risk management documentation to form a holistic picture.
This article is part of the Virtual MedTech: EUMDR exchange conference, 2021 report. Get your full copy of this comprehensive report today!
Cactus Life Sciences
Cactus Life Sciences is a medical communication company that provides scientific strategy and content across the healthcare continuum, anywhere in the world ─ with a focus on science, innovation, and efficiency. We work alongside leading healthcare companies to establish the optimal role of medicines and encourage positive behaviors (physician and patient) that improve patient outcomes.